It's caused by insulin resistance! But which one? The first, the second, or even the third?
Interestingly, most experts in the field of civilization diseases have not yet even taken note of the opinion of scientists who see and prove that the first well-measurable manifestation of metabolic problems is insulin resistance. It is manifested by an increased level of insulin in the blood after eating and on an empty stomach. However, if we are to find the cause of insulin resistance, we will get into trouble.
Among the supporters of the so-called ketogenic diet, there is a belief that the cause is carbohydrates and sugars in food. Just leave them out and the insulin level will drop. They consider this to be sufficient evidence. Carbohydrates and sugars are poisons. Done, insulin resistance solved. What does matter is that a low-carb ketogenic diet temporarily disables insulin production so that, for example, a standard oral glucose tolerance test will be unsatisfactory, similar to a diabetic. Isn't that insulin resistance? As Dr. Bikman rightly says, it is not. It is a temporary carbohydrate intolerance. For a successful test, you need to eat at least 150 g of carbohydrates a day for several days before the test! Or that the standard diet of rodents is a low-fat diet containing up to 80% carbohydrates? And it doesn't cause them any metabolic problems! Carbohydrates simply don't cause insulin resistance.
I have already discussed the issue of insulin resistance several times here. It turned out that there are two types of insulin resistance.
The first, immediate, is a manifestation of competition between different fuels that cannot enter the mitochondria at the same time. One fuel gets priority and the other fuels simply have to wait. It is very clearly observable when consuming alcohol, but it is also seen when burning fat. Alcohol has priority over all other fuels. Similarly, fats have priority over glucose. So, if we consume them at the same time, glucose can be stored in glycogen, but it cannot be burned. This immediately raises insulin levels. I call this phenomenon metabolic insulin resistance, and it is a completely positive phenomenon because it protects the mitochondria from overloading and from switching the metabolism into emergency mode, into senescence. I used to call this emergency mode pseudohypoxia, but the more general name used in scientific studies is now cellular senescence.
 |
| Immediate insulin resistance caused by alcohol (ethanol) metabolism will be manifested by increased insulin levels during glucose infusion. From the first minute! |
A study on a mouse model was published that explains mechanistically what happens during a short-term (one week) overeating of a high-fat diet. The main finding is that insulin resistance occurs in adipose tissue. The concentration of a specific type of fat, diacylglycerols (DAG), increases in cell membranes. We already know triacylglycerols (triglycerides, TAG). Three fatty acids are linked to glycerol. Diacylglycerols have only two fatty acids and activate processes that lead to the deactivation of insulin receptors.
So we have the first protection, which appears to be insulin resistance, which works from the first minute, simply immediately when needed. It does not deactivate insulin receptors, but it increases insulin levels and suppresses the entry of glucose into the cell. Its activation is beneficial, and insufficient activation leads to an overload of mitochondria when there is an excess of fuel.
The second is a slightly delayed insulin resistance, caused by the accumulation of DAG in membranes and, to my knowledge, is also beneficial. It also prevents the entry of excess fuel, this time only glucose, into the cell by suppressing the function of insulin receptors. With insufficient activation of insulin resistance, the storage of unburned fuel into triglycerides, into fat droplets, is activated. This probably also causes higher production of DAG and activation of the above-described mechanism of insulin receptor deactivation, which is so surprising to some scientists. They do not understand at all that insulin resistance could be beneficial. That it is a protective mechanism programmed by evolution.
Excess fuel also activates the process of de novo lipogenesis (DNL), and there is a high probability that in some cells, with a certain fat composition, the maximum safe consumption limit of NADPH will be exceeded. This leads to insufficient antioxidant protection by glutathione; hydrogen peroxide is not neutralized and its excess activates the enzyme PLA2, which then releases signaling molecules from the phospholipids of the mitochondrial membrane, so-called oxylipins.
 |
| Beta oxidation of fats produces superoxide O2- and H2O2, which activates UCP2 via PLA2 and membrane phospholipids, but only if fat formation is not activated by PPARγ. |
 |
| Oxygen consumption and decrease in inner mitochondrial membrane potential during palmitate burning. Oxidation is strongly promoted through PLA2/UCP2 activation. Their deactivation (KO) almost stops oxidation. |
And now it gets interesting because these oxylipins can trigger either mitochondrial uncoupling (lean phenotype) or senescence (obese phenotype). If the process of creating new fat DNL were not activated, then mitochondrial uncoupling (lean phenotype) would win, heat production would increase and the efficiency of ATP production would decrease. We would simply burn more fuel than is currently needed. But we know that the DNL process has already been triggered precisely because of the need to get rid of excess glucose by storing it in new fat, so oxylipins activate the PPARγ factor, and it decides to trigger cellular senescence (obese phenotype). Even different membrane compositions (food history) can lead to this outcome. In order not to trigger senescence, we would have to ensure the suppression of PPARγ by turning off pseudohypoxia, turning off the HIF1 factor, and activating AMPK. As we already know, short-chain fatty acids, especially acetate/vinegar produced by intestinal bacteria, can do this.
 |
| The main limiting factor is mitochondrial NADPH production by the enzyme IDH2, the amount of proteins is the same.... |
 |
| ...but mRNA expression, i.e. the requirement for the production of the IDH2 enzyme, is much higher, so there is not enough for proper function. That much NADPH cannot be produced... |
 |
| ... which causes senescence activation in some fat cells, leading to insulin resistance and weight gain. |
What will genetically disabling IDH2 cause, i.e. creating NADPH deficiency?
 |
| A high-fat diet causes obesity, and IDH2 is already working beyond its limits. Genetically turning off IDH2 leads to even faster weight gain. |
What if we turn off senescence?
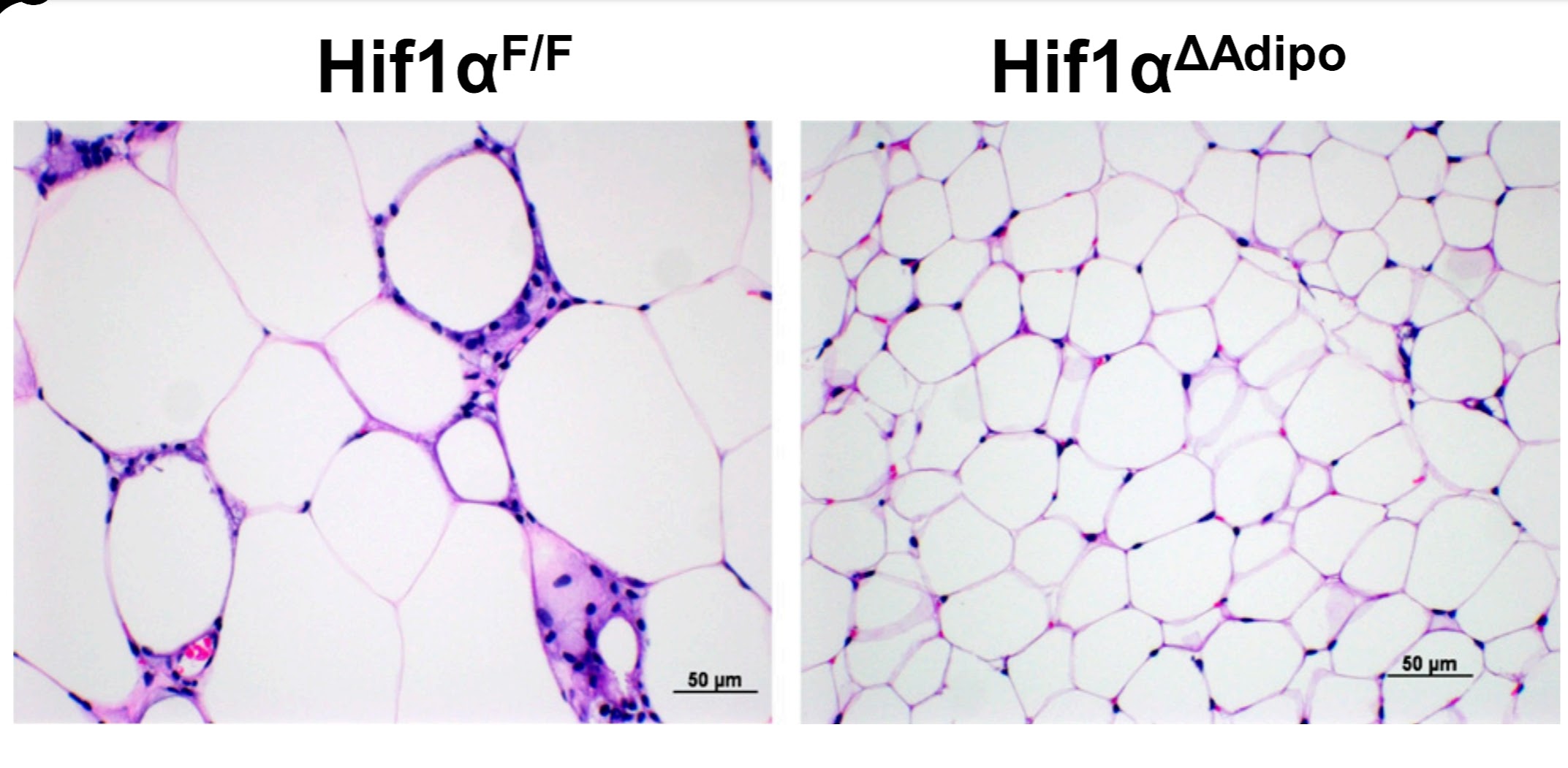 |
| Turning off the transcription factor HIF1A selectively in fat cells completely prevents cellular senescence and insulin resistance, resulting in the absence of hypertrophy, and fat cells remaining small and fully functional. |
If we trigger senescence, cells send chemical signals (SASP factors) that cause insulin resistance. In addition to the original two positive insulin resistances, we now have a third, the only one with negative effects. The two previous ones have positive effects on metabolism because they prevent the third variant from being triggered when senescent cells call on immune system cells and cause chronic inflammation. This process was originally triggered as a protection against excessive consumption of NADPH molecules, but the immune system, on the contrary, increases NADPH consumption by activating the NADPH oxidase NOX2. Cellular senescence is a permanent state for which we do not yet know how to turn it off. This can only be achieved by completely removing senescent cells through apoptosis or ferroptosis. Selective removal is basically harmless because there are not too many senescent cells.
 |
| Senescent cells in human adipose tissue are characterized by activated p21 and, when implanted into the adipose tissue of mice, cause insulin resistance (V). Selective removal of senescent cells in the implant suppresses this effect (DQ). |
If we think about the original problem of the lack of NADPH for fat formation, the easiest way would be to suppress the formation of new fats (DNL) in an environment where there is enough fat. We have already encountered this, it is a consequence of the simultaneous burning of glucose and fats, which also adds glutamine as the main raw material for the formation of new fats. Phosphorylation of the enzyme ACC, i.e. activation of the enzyme AMPK, will help.
To summarize, insulin resistance, which prevents glucose from entering the cell, has a protective effect in most cases. It prevents the cell from overloading with fuel, reduces the production of H2O2, and, therefore, inhibits the activation of the enzyme PLA2. It thus suppresses the signaling that, depending on the activity of the PPARγ factor, either triggers mitochondrial uncoupling or cellular senescence.
There is only one case where insulin resistance hurts metabolism. This is when some fat cells activate senescence and thus trigger insulin resistance and inflammation in the surrounding fat cells, manifested by NOX2 activation and increased H2O2 production. Thus, senescence ensures the permanent activation of insulin resistance, which is no longer beneficial and leads to further metabolic disorders.
References:
Ethanol causes acute inhibition of carbohydrate, fat, and protein oxidation and insulin resistance
Inhibiting HIF-1 signaling alleviates HTRA1-induced RPE senescence in retinal degeneration
Oxylipin-PPARγ-initiated adipocyte senescence propagates secondary senescence in the bone marrow
Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity



I was thinking about how NADPH is a cofactor in the reaction facilitated by the enzyme stearoyl-coa-desaturase-1 (SCD1), which helps convert saturated fat to monounsaturated fat. Do you think NADPH depletion is why high saturated fat diets in animal studies cause metabolic issues? We know that improper cell membrane fluidity leads to endoplasmic reticulum stress, which damages the cell, and these high-fat mouse diets are lacking in nutrients. So I'm wondering if that lack of nutrients weakens antioxidant defense, meaning more NADPH is needed to recycle glutathione, leaving insufficient NADPH for the SCD1 reaction, therefore leaving cell membranes too stiff in the context of a high saturated fat diet? Especially because high-fat diets containing mostly monounsaturated fat don't seem to cause the same issues in these mice. But I'm thinking with proper nutrition, saturated fat is harmless.
ReplyDeleteGreat point. In reality saturated fats are accompanied with medium and short-chain fats, these are protective, they activate omega oxidation to dicarboxylic acids. They are processed in peroxisomes and make acetate. Similar effect as adding fish oil, DHA. It's pity that acetate is not evaluated as one of the most important modulators of fat metabolism.
ReplyDelete