Can Vinegar Be the Solution to Alzheimer's and Senile Dementia?
Vinegar or acetate is a very interesting substance, a food supplement. If you would like to know more and in detail about it and its metabolism in the body, read this two-part article. It is devoted to acetate only and has two parts, a total of 49 densely annotated pages in two columns. All this about a molecule that contains only two carbons. Incredible. It apparently affects all processes in the body.
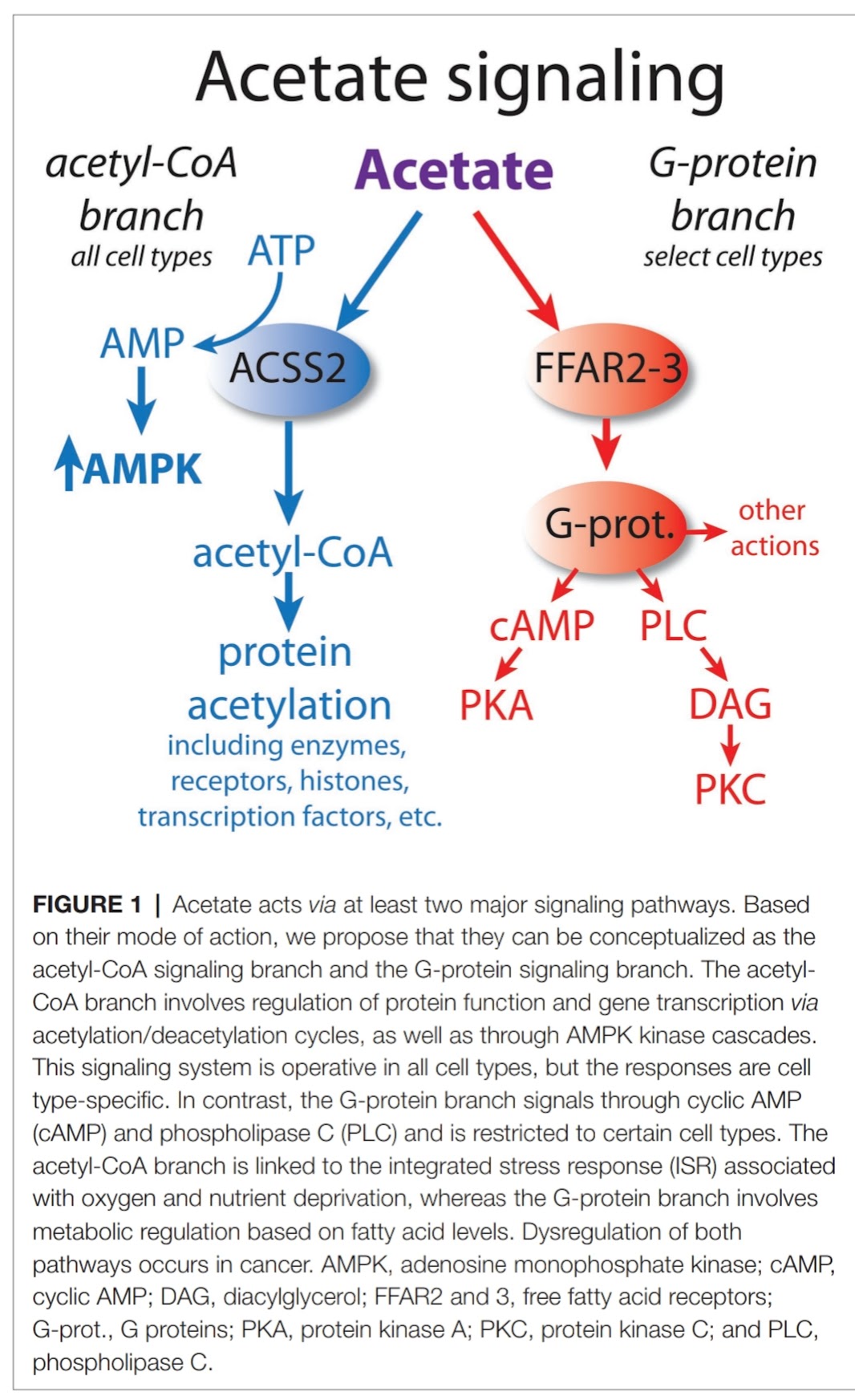 |
| Acetate acts not only by activating AMPK during its conversion to acetyl-CoA, but also directly on the FFAR2 receptor, thus, for example, it supports vasodilation, i.e. better blood and oxygen supply to tissues. |
First, let's review the basic mechanism by which acetate in the body acts against obesity. It is the result of the processes associated with the activation of acetate in the cell, i.e. the connection with the CoA molecule to form acetyl-CoA. This reaction consumes energy (ATP) and creates a signal of shortage, activates the enzyme AMPK, which limits the ACC enzyme by phosphorylating it to pACC. This is probably the most important change in the fight against obesity. Another no less important effect is the reduction of lactate formation in connection with greater CO2 release by supporting the activity of the TCA cycle. As I mentioned before, carbon from food is either built into fat cells or is exhaled as CO2.

It is the activity of the ACC1 enzyme that serves as a switch between storing carbon in adipose tissue or releasing CO2 through the lungs. As we age, more fat cells switch from oxidative to fermentative metabolism, without the use of oxygen. It can also affect cells in other tissues, possibly even in the brain. Fermenting cells gradually poison the environment of healthy energy-producing cells (ATP) by oxidation. The causes can be various, insufficient blood supply to certain tissues, slowed activity of the electron transport chain in the mitochondria, various poisonings, etc.
We know that cells in a high-fat environment should never switch to anaerobic mode, which does not use oxygen. Fats simply cannot be fermented, this can only be done with glucose. The switch occurs by accumulating succinate in the cell and stopping the degradation of the transcription factor HIF-1α. In an environment where long fatty acids are processed, there is a lack of intermediates for the accumulation of succinate, so the threshold for HIF-1α activation is much higher than in cells with a low fat content. This differentiates tissues, in some tissues HIF-1α switching is desirable, in others it is devastating

Now let's go back to acetate and its ability to activate the AMPK enzyme and shut down the ACC1 enzyme, thus closing the path of carbon to the formation of new fats. This has yet another significant effect, the concentration of acetyl-CoA molecules in the cytoplasma, where the HIF-1α transcription factor is also found, will increase. And so its acetylation occurs. While acetylation in the mitochondria means slowing down activity, acetylation in the cytoplasma has a completely different meaning, it is used in the quality control of proteins during their formation in organelles called the endoplastic reticulum (ER).
The formation of new proteins takes place by assembling a chain of amino acids according to the sequence of nucleotides in RNA. However, this cord does not yet function as an enzyme. In order to assemble a new nanomachine, which we call an enzyme, it must be folded correctly, a complex and vulnerable process. If it fails, the newly formed protein is not acetylated enough during the output control, so it differs from the correctly assembled protein, which is highly acetylated. Upon exiting the checkpoint, the acetylated correctly assembled protein is deacetylated and released. Low acetylated protein is broken down.
Newly formed enzymes are not acetylated. The newly formed HIF-1α is not acetylated and serves as a so-called watchdog circuit, i.e. a circuit that monitors whether the system is working. If the system works, it can immediately destroy HIF-1α. If this fails, the system has a problem and needs to go into repair mode, the electronic system usually reboots. The biological system switches to backup metabolism, fermentation.
So far, only the hydroxylation system with subsequent removal is reported in the literature. But I found a study that shows that the main mechanism for normoxia is acetylation with subsequent degradation of HIF-1α. This is important information in the context of the fact that stopping the activity of ACC1 stops the uptake of acetyl-CoA to fat synthesis, elevates it's level and thus increases the acetylation of HIF-1α, ensures its degradation, and also ensures the proper acetylation of histones to properly control gene activation in the cell nucleus. In addition, stopping the formation of fat will significantly reduce the consumption of NADPH. In a glucose-deficient, fat-excess environment, this means that NADPH generation by the cytosolic enzyme IDH can be stopped, which fixes the reversal run of the mitochondrial IDH enzyme which blocks the TCA cycle. So the TCA cycle starts to run clockwise and can produce CO2. This will provide enough oxygen in exchange for CO2. Are you watching? This is the correction of oxidative metabolism, oxidative phosphorylation. Acetate/vinegar can do that.
Now I'm going to leave the acetate and look at a mouse model of Alzheimer's disease. We know that the current treatment procedure for "Alzheimer's" is completely ineffective and there is no explanation of the causes of this increasingly common disease. This group of scientists took the opposite approach, first testing a number of substances in a mouse model and conducting a study on the two most promising. They seem to have succeeded. They also determined the mechanism of how two completely different synthetic substances act on the brain in such a way that they restore the metabolic parameters and cognitive abilities of mice.

And if you take a closer look at the mechanism, wow, it's exactly what I described in relation to acetate and its effect on fat cells. Would there be a connection? The effect of acetate is dependent on the activity of ACSS2 enzyme, it is most active in adipose tissue. It's not measured to be very active in the brain, but since adipose tissue sends signals to the body, reprogramming of fat cells could also allow distant brain support cells to switch from fermentation to oxidative phosphorylation. Is it at all possible that Alzheimer's disease would have the same cause as obesity, the switching of part of the cells from oxidative phosphorylation to fermentation, i.e. aerobic glycolysis, i.e. activation of HIF-1α?

Let's take a closer look at the results of measuring succinate levels, a HIF-1α activator. At first it is low (9mths), with advancing Alzheimer's disease its level increases (13mths). Also watch the spread of the values. While the other intermediates have a relatively small variance (except for α-ketoglutarate, the precursor of succinate), it is different for succinate, it resembles a mixture of YES/NO states. We already know that the switching of HIF-1α takes place as a switch in connection with the activity of NOX2 enzymes.
HIF-1α activation turns on H2O2 production by NOX2, and H2O2 production turns on HIF-1α. Some cells therefore ferment and burden the environment with oxidative stress from NOX2. Others use oxygen for oxidative phosphorylation, but are disrupted by ambient H2O2. Hydrogen peroxide oxidizes polyunsaturated fats in membranes. How will it manifest itself? Observe the level of omega-6 linoleic acid (Linoleyl carnitine) in the following image. Why do levels of linoleic acid decrease with advancing dementia? As the membranes are damaged, they must also be repaired and replenished, so linoleic acid is disposed more.

But we also see an increase in linoleic fatty acid after treatment application (13mths+CMS121, 13mths+J147). The treatment knocked down succinate levels, so no fermentation, no HIF-1α nor NOX2 activation, substantially reduced oxidative stress, and no membrane destruction. Everything fits together. The same process that I have shown in adipose tissue also occurs in brain tissue to cells that provide nutrition and support to nerve cells. Dementia and Alzheimer's disease is caused by the activation of pseudohypoxia and we know that omega-6 linoleic acid in particular enables the activation of HIF-1α in an environment where it should not occur, in fat cells. Now we can also add brain cells to this.
Make no mistake that supplementing with polyunsaturated fatty acids, such as omega-3, can solve the problem. It will only provide fresh material for the creation of more poisons that will arise from damaged membranes. The solution is to revert the metabolic state back to oxidative phosphorylation. We already know that acetate can do this in mice, maybe with the right application it will be able to do it in humans too, maybe it can even change the course of Alzheimer's disease and senile dementia, who knows.
Acetate is also a product of ketone metabolism, so the effect of a ketogenic diet suppressing e.g. epilepsy in children could be a result of acetate metabolism. Similarly, the positive effect of a small amount of alcohol is most likely the result of the action of acetate. And finally, we can mention the metabolism of alpha linolenic acid (C18:3 n-3), which strongly activates acetate production by intestinal bacteria, so that unlike long omega-3, the positive effect of acetate outweighs the negative effects.
 |
| The main component of coconut oil C12:0 as protection against insulin resistance, the main component of olive oil C18:1n-9 is neutral but metabolic products of omega-6 linoleic acid from other vegetable oils (C20:3n-6, C20:4n-6, C22 :4n-6) definitely do not help. Even long omega-3 fats do not help. Of the omega-3s, only ALA (C18:3 n-3) has a positive effect (likely via acetate produced by the gut microbiome). |
And an added bonus, triacetin really works against dementia in a mouse model.
References:
Causes and Consequences of A Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism
Elevating acetyl-CoA levels reduces aspects of brain aging
Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1
Adipose tissue fatty acids and insulin sensitivity in elderly men
NADPH—The Forgotten Reducing Equivalent
ACSS2-dependent histone acetylation improves cognition in mouse model of Alzheimer’s disease



Comments
Post a Comment