Deactivation of the ARNT/HIF1β signal can prevent diabetes (T2DM) in mice
Since the very first post, I have tried to find the reason why the percentage of type 2 diabetics is constantly increasing in today's society. One of these analyzed scientific studies with the aim of finding the etiology of this disease, i.e. the real cause. If I had to summarize it briefly, at first, the fat cells are reprogrammed to release more free long-chain fatty acids (FFA) into the circulation, thus overloading the other organs with excess fuel. Lack of short and medium fatty acids will slow down metabolism and reduce fat burning. This causes a further increase in fatty acids.
This increased level of FFA activates higher insulin secretion, but only initially. As demands for high insulin production increase, insulin resistance worsens, insulin production begins to gradually decrease, and glucose levels increase. This starts to damage other organs. The question is, are the beta cells of the pancreas damaged or are they just reacting to the metabolic state with a defense mechanism that includes suppression of insulin secretion? There is no clear answer to this yet.
Let's take a closer look at one mouse study that shows that even on a high-fat, high-sugar (HF) diet, deactivating of the ARNT signal, also known as HIF1β, can prevent HIF1α and HIF1β parts from joining, thus turning off pseudohypoxia and that way preserving metabolic flexibility and low insulin resistance. But it won't prevent changes in adipose tissue. Mice with active HIF1β (+/+/Cre) were exactly as fat as mice with HIF1β turned off (denoted as fl/fl/Cre or my note KO).
What is ARNT/HIF1β? It is one part of the HIF1 transcription factor. This switches the cell into rescue mode in the event of a lack of oxygen and turns on glucose fermentation. But in a fatty environment with a lack of glucose (between meals, at night) HIF1 should never be activated, fats cannot be fermented. It is provided on the one hand by the lack of intermediate products of glucose metabolism. But it is not the only mechanism, ARNT is part of the carbohydrate detection system. It should be deactivated when there is a lack of carbohydrates. However, this is not the case in the current toxic environment. ARNT also detects substances such as dioxin. It can also be activated by other toxins. This could be one of the causes of the increasing number of diabetes cases.
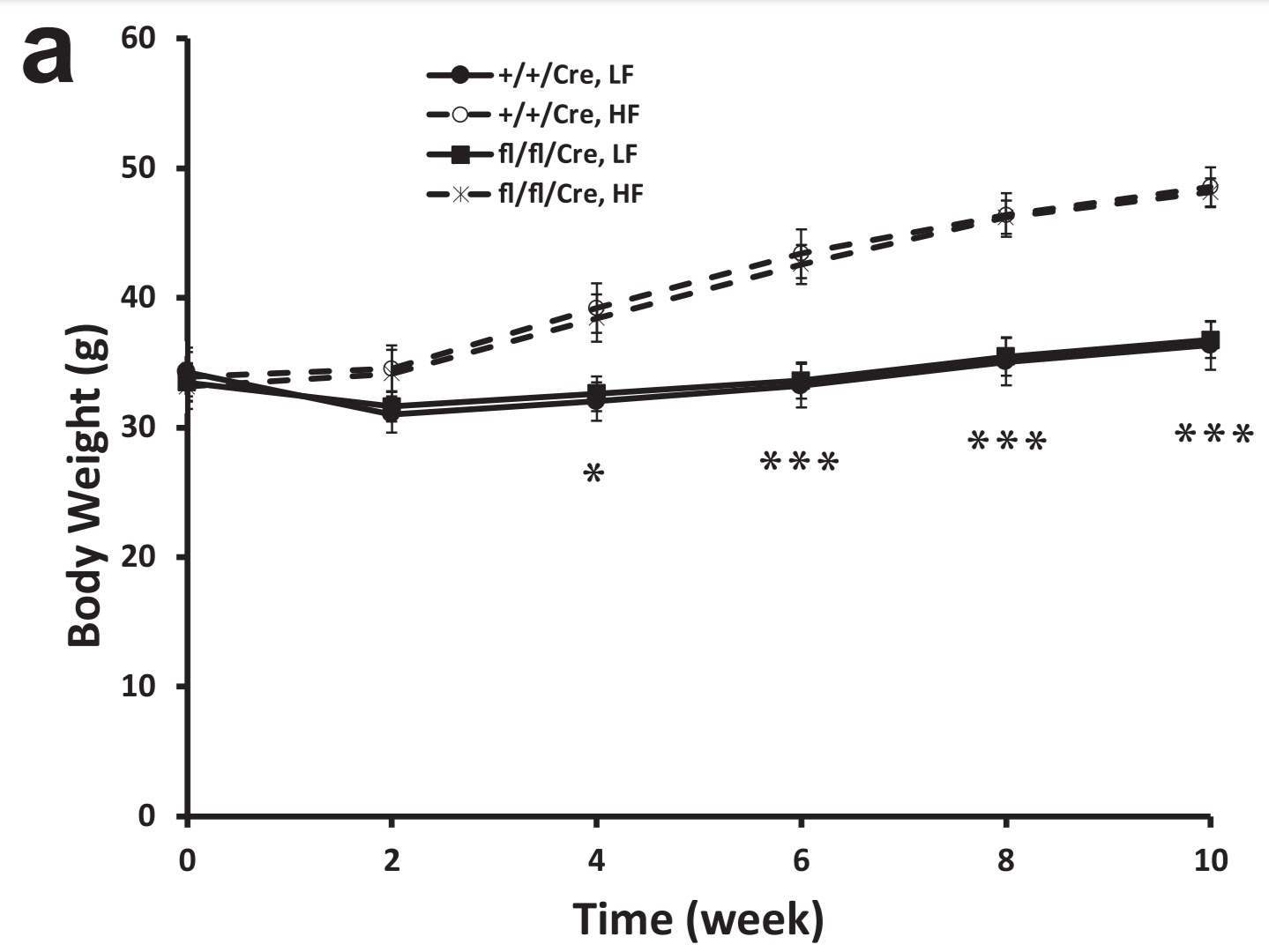 |
| Deactivation of pancreatic HIF1β does not affect obesity on a high-fat diet with sugar (fl/fl/Cre, HF). |
 |
| Deactivation of pancreatic HIF1β normalizes insulin resistance under a high-fat diet with sugar (fl/fl/Cre, HF). |
 |
| Glucose AUC (area under the curve) is normalized by inactivating pancreatic HIF1β. |
 |
Metabolic flexibility is normalized by pancreatic HIF1β deactivation. Without deactivation, a fatty diet with sugar activates pseudohypoxia, reducing fat metabolism and overall metabolic rate. |
 |
| Note that the normal response to an increase in free fatty acids (FFA) is increased insulin secretion. Activation of pseudohypoxia by a high-fat diet with sugar reverses the response and reduces insulin secretion while increasing FFA. Insulin reduces lipolysis, and its reduction therefore leads to a further increase in FFA, creating a vicious circle that leads to the suppression of insulin secretion and a large increase in FFA, i.e. type 2 diabetes. Knockdown of ARNT/HIF1β restores the normal beta cell response to FFA elevation. |
Check out this older post. In it, we find information that insulin secretion has at least three components and is dependent not only on glucose levels, but also on activation by fatty acids or hydrogen peroxide. It's not a simple relation at all. Insulin not only regulates the level of glucose, but also lipolysis, i.e. the level of free long fatty acids. For proper regulation, a positive slope of the dependence of insulin secretion on the FFA level must be maintained. If the relation is reversed by high glucose and/or FFA levels, regulation stops working and type 2 diabetes develops. This can be prevented by turning off pseudohypoxia in the beta cells of the pancreas. So, if switching off pseudohypoxia is preventive, could vinegar (sodium acetate, triacetin) restore beta cell function via switching off the transcription factor HIF1?
Addendum
Sodium acetate appears to work against diabetes in the STZ rat model. So everything is as I predicted in the previous paragraph. Sodium acetate suppresses de novo lipogenesis by activating AMPK and phosphorylating ACC1. This is manifested by a decrease in deamination, normal clockwise run of the TCA cycle, and a decrease in the formation of uric acid by xanthine oxidase. But the authors of this study do not know about it.
 |
| Sodium acetate (SAT) significantly reduces fatty liver in a model of diabetes. |
 |
| Sodium acetate (SAT) reduces oxidative stress and increases levels of the major antioxidant GSH. |
 |
| Sodium acetate (SAT) reduces uric acid levels and inflammation. |
References:
Fatty Acids Prevent Hypoxia-Inducible Factor-1a Signaling Through Decreased Succinate in Diabetes



Comments
Post a Comment